

使用单细胞测序分析软件Cell Ranger进行测序文件的处理
使用Cell Ranger处理10x Genomics测序数据是一个多步骤的过程,涉及到数据的质量控制、读段的映射到参考基因组、识别和定量单细胞中的转录本。
- 5’端(五端):指一个核酸分子中磷酸基团连接的那一端。在DNA和RNA的线性表示中,5’端通常位于左侧。
- 3’端(三端):指一个核酸分子中羟基连接的那一端。在DNA和RNA的线性表示中,3’端通常位于右侧。
检查FASTQ文件
Cell Ranger进行测序的FASTQ文件通常遵循特定的命名约定:
sample_S1_L001_R1_001.fastq.gz
sample_S1_L001_R2_001.fastq.gz
sample_S1_L002_R1_001.fastq.gz
sample_S1_L002_R2_001.fastq.gz
命名规则
[Sample Name]_S1_L00[Lane Number] _[Read Type]_001.fastq.gz
这里,L001和L002表示流水线编号,R1和R2表示读段的方向。
- I1: Sample index read (optional)
- I2: Sample index read (optional)
- R1: Read 1
- R2: Read 2
运行Cell Ranger count
cellranger count --id=MyProject \
--transcriptome=/path/to/reference_genome \
--fastqs=/path/to/my/fastq_files \
--sample=sample
–id=MyProject:指定输出目录的名称,Cell Ranger会在当前目录下创建一个名为MyProject的文件夹,其中包含运行结果。
–transcriptome=/path/to/reference_genome:指向你的参考基因组的路径。这应该是使用cellranger mkref创建或下载的参考基因组的文件夹。
–fastqs=/path/to/my/fastq_files:包含你的FASTQ文件的目录路径。
–sample=sample:指定FASTQ文件名中共有的样本名前缀。在这个例子中,所有文件都以sample开头,因此使用sample作为参数。
对于多个样本的分析,可以使用cellranger multi命令,它允许在一个运行中处理多个样本,并可以进行多种类型的分析,如单细胞转录组学和单细胞ATAC-seq的联合分析。
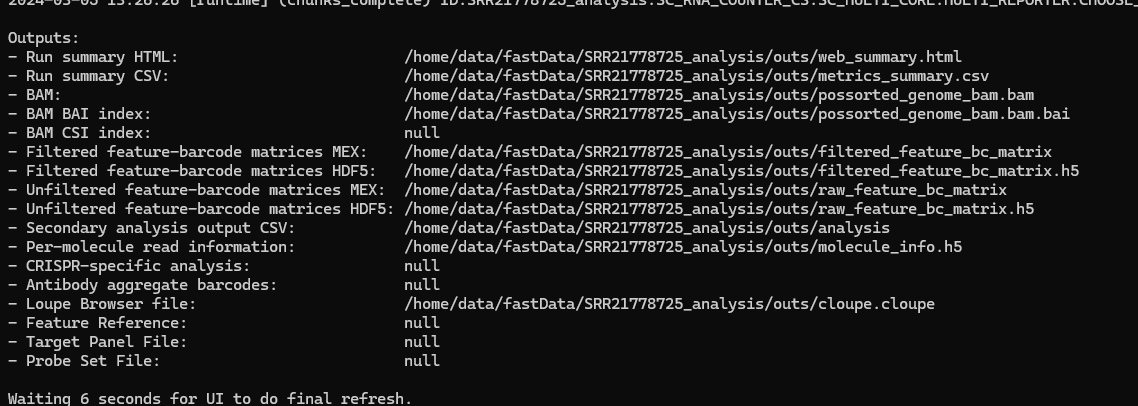
Cell Ranger运行完成后,会生成一个包含多个文件和文件夹的输出目录。重要的输出包括:
web_summary.html:一个包含高级别质量控制指标的可视化报告,可以在浏览器中查看。outs/filtered_feature_bc_matrix/:包含过滤后的基因表达矩阵,可用于后续的分析,如聚类、差异表达分析等。
例子
# 压缩
$ gzip /Sample/Sample1.fastq
$ gzip /Sample/Sample2.fastq
# 改名
$ mv Sample1.fastq.gz Sample1_S1_L001_R1_001.fastq.gz
$ mv Sample2.fastq.gz Sample2_S1_L001_R1_001.fastq.gz
# 运行
nohup cellranger count --id=Sample_analysis --transcriptome=/home/data/GenomeLib/refdata-gex-mm10-2020-A --fastqs=/home/data/fastData/Sample --localcores=10 &
- –id:指定输出文件存放目录(我这里的是样本名)
- –localcores :线程数
- –transcriptome :参考基因组文件目录,里面含有构建好的index
- –fastqs :fastq文件所在路径
- –sample:要和fastq文件的前缀中的sample保持一致,作为软件识别的标志
然后查看 nohup 文件看日志, 就开始验证运行了
Martian Runtime - v4.0.11
Serving UI at http://server:33767?auth=jWMOT6C3fxc34jtIjYJLr2zv_a7hjA7RxgLQU-Bbvvw
Running preflight checks (please wait)...
Checking sample info...
Checking FASTQ folder...
Checking reference...
Checking reference_path (/home/data/GenomeLib/refdata-gex-mm10-2020-A) on kjtjserver...
Checking optional arguments...
结果
注意
- 在使用
--sample参数时,需要确保指定的样本名与FASTQ文件名中的样本名完全匹配。如果样本名指定错误,Cell Ranger将无法识别正确的文件。 - Cell Ranger期望输入的FASTQ文件遵循特定的命名约定,如
SampleName_S1_L001_R1_001.fastq.gz。这里SampleName应与--sample参数中指定的样本名相匹配,S1表示样本编号,L001表示流水线号,R1或R2表示读段方向,001是分割文件的编号。如果你的文件名不遵循这个约定,Cell Ranger将无法正确识别它们。 - 使用
--fastqs参数指定的目录应该直接包含FASTQ文件或FASTQ文件所在的子目录。如果指定的目录不正确,或者FASTQ文件不在该目录下,Cell Ranger将找不到这些文件。 - 如果你的测序数据来自特定的测序通道,需要通过
--lanes参数指定这些通道。如果指定错误,或者不需要此参数但却错误地使用了它,可能会导致Cell Ranger找不到匹配的FASTQ文件。 - Cell Ranger在处理数据时预期至少有
Read1和Read2。