

官网: https://cole-trapnell-lab.github.io/monocle3/
为什么要做轨迹分析以及轨迹分析的定义
在进行标准的单细胞分析流程得到聚类结果后,可以进行轨迹分析,并进一步进行基因表达分析。
但并不是只有聚类结果才可进行轨迹分析,标准分析中的很多步骤都可以follow with轨迹分析。
在整个生命生长发育过程中,细胞都在不断从一种功能“状态”过渡到另一种功能“状态”(如下图)。处于不同状态的细胞表达不同的基因,产生蛋白质和代谢物的动态重复,从而完成它们的工作。当细胞在不同的状态间转化时,会经历转录重组的过程,一些基因被沉默,而另一些则被激活。这些瞬态通常很难描述,因为在更稳定的端点状态之间净化细胞可能很困难或不可能。但由于这个过程是连续发生的,我们可以使用轨迹推断(TI,trajectory inference)的方法可以根据测序的细胞(瞬时状态)之间表达模式的相似性对单细胞沿着轨迹进行排序,以此来模拟细胞动态变化的过程。也就是重建分化轨迹或者拟时间轴。
安装
先安装依赖
BiocManager::install(c('BiocGenerics', 'DelayedArray', 'DelayedMatrixStats','limma', 'lme4', 'S4Vectors', 'SingleCellExperiment','SummarizedExperiment','batchelor', 'HDF5Array','terra', 'ggrastr'))
然后再安装monocle3
install.packages("devtools")
devtools::install_github('cole-trapnell-lab/leidenbase')
devtools::install_github('cole-trapnell-lab/monocle3')
举个粒子
# 1.创建CDS对象
data <- seurat_obj@assays$RNA@counts
cell_metadata <- seurat_obj@meta.data
gene_annotation <- data.frame(gene_short_name = rownames(data))
# 2.转换 Seurat 对象为 Monocle3 对象
cds <- new_cell_data_set(data,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
## 3.预处理和降维
cds <- preprocess_cds(cds, num_dim = 50) # 预处理
cds <- reduce_dimension(cds) # 降维
## 4.构建时序图
cds <- cluster_cells(cds) # 聚类
cds <- learn_graph(cds) # 识别轨迹
## 5.画图
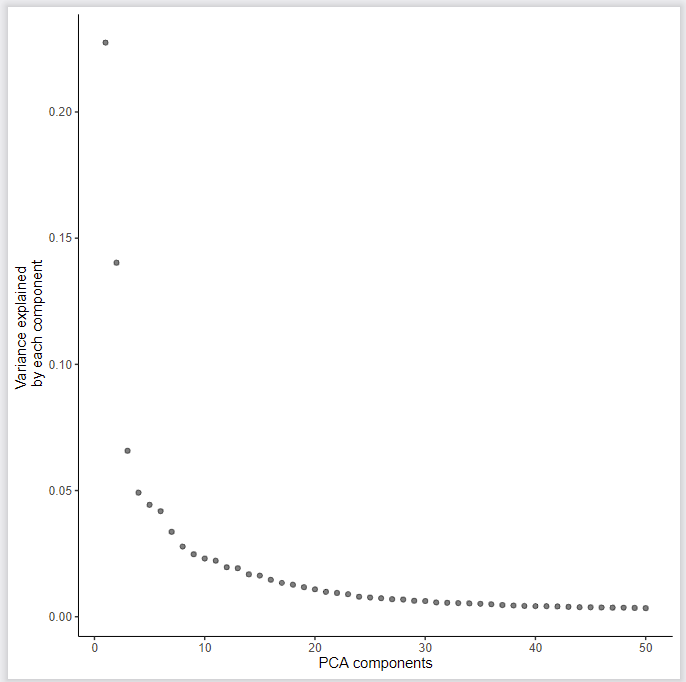
plot_pc_variance_explained(cds) # 很像Seurat的ElbowPlot()
plot_cells(cds, color_cells_by = "partition", label_groups_by_cluster = FALSE) # 进行可视化
preprocess_cds 详解:
- 降维方法method = c(“PCA”, “LSI”),默认是PCA
- 默认维度num_dim是50
- 初步降维前归一化norm_method 默认是log处理
- 设置降维方法是PCA时,自动进行标准化处理
导入整合过的umap坐标后作图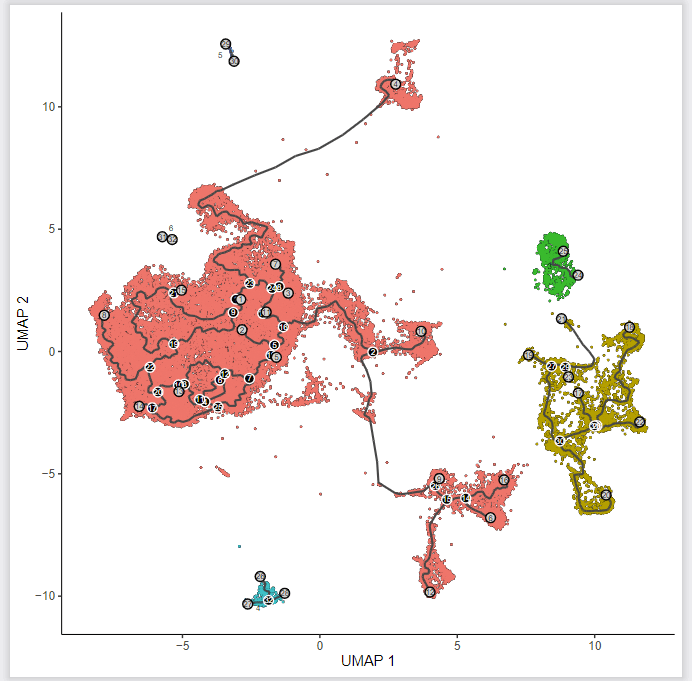
目的
1.寻找拟时轨迹差异基因
拟时轨迹是指在发育过程中细胞状态随时间(或类似指标)的变化。通过寻找拟时轨迹差异基因,我们可以找到在不同发育阶段或状态中表达水平有显著变化的基因。这些差异基因可能在细胞发育和分化过程中扮演重要的角色,例如调控特定发育途径或细胞类型的标志物。拟时轨迹差异基因的鉴定有助于理解细胞发育过程中的基因调控和细胞状态转换。
2.空间共表达效应
空间共表达效应是指在空间中距离相近的细胞之间基因表达的相似性。在组织或器官中,相邻的细胞可能具有类似的功能和特性,因此它们的基因表达也可能有相似之处。通过研究空间共表达效应,我们可以揭示细胞在空间上的组织结构和相互作用模式。这有助于了解细胞在组织中的空间分布、细胞-细胞相互作用以及细胞特异性的功能。
通过寻找拟时轨迹差异基因和研究空间共表达效应,我们可以深入了解单细胞数据中的动态细胞状态和空间结构,为细胞发育和组织功能的研究提供重要线索。
作图扩展
根据基因表达量进行映射
plot_cells(cds, genes=c("cpna-2", "egl-21", "ram-2", "inos-1"))
如果使用tSNE方法降维的话:
cds <- reduce_dimension(cds, reduction_method="tSNE")
# 然后对tSNE结果可视化
plot_cells(cds, reduction_method="tSNE", color_cells_by="cao_cell_type")
用多了再加细节。