什么是富集分析
一个生物过程通常是由一组基因共同参与,而不是由单个基因独自完成。富集分析的基本前提假设是,如果一个生物学过程在已知的研究中发生异常,则共同发挥功能的基因极可能被选择出来作为一个与这一过程相关的基因集合。
富集分析的几种类型
1.GO富集分析
GO富集分析会从三个方面描述基因潜在的功能
分子功能(Molecular Function,MF)—— 即基因是否富集到分子相关的通路上细胞组分(Cellular Component,CC)—— 即基因定位在细胞的哪个位置上参与的生物过程(Biological Process,BP)—— 即基因参与哪些生物学过程
离子通道活性的GO term是GO:0005216,如果差异基因富集到该term上,那么所研究的基因可能与离子通道的激活与抑制有关联。
2.KEGG富集分析
京都基因与基因组百科全书(KEGG)是了解高级功能和生物系统(如细胞、生物和生态系统)、用于研究通路的数据库之一。KEGG 通路分析是借助 KEGG 数据库(Kyoto Encyclopedia of Genes and Genomes),对所有鉴定到的基因进行通路注释,并分析这些基因参与的主要代谢和信号转导途径。
简单来说,使用KEGG数据库中通路的注释信息,将基因与已知的代谢通路和功能进行关联
富集分析步骤
我们使用 clusterProfiler 来进行富集分析
GitHub地址: https://github.com/YuLab-SMU/clusterProfiler
# 下载包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")
# 加载包
library(clusterProfiler)
library(org.Mm.eg.db) # 鼠鼠的基因库
library(dplyr)
# 读取分群聚类之后的结果
all_markers <- read.csv(file = "./data/all_markers_res02.csv")
# 提取特定亚群的显著差异表达基因()
for (i in 1:15) {
subgroup_de_genes <- all_markers %>%
filter(cluster == i - 1 & p_val_adj < 1e-50 & avg_log2FC > 0.6) %>%
pull(gene)
head(subgroup_de_genes)
# 进行GO富集分析
enrich_result <- enrichGO(gene = my_genes, # 需要进行富集分析的基因列表,通常是一组差异表达基因或特定基因集
OrgDb = "org.Mm.eg.db", # 根据你的物种选择合适的数据库 org.Hs.eg.db(人类) org.Mm.eg.db (鼠鼠)
keyType = "SYMBOL", # 基因列表中使用的标识符类型,如"SYMBOL"(基因符号)、"ENTREZID"(Entrez基因ID)、"ENSEMBL"(ENSEMBL基因ID)等
ont = "BP", # 指定要分析的GO本体类别,可以是"BP"(生物学过程)、"CC"(细胞组分)或"MF"(分子功能)
pAdjustMethod = "BH", # 用于多重检验校正的方法,如"BH"(Benjamini-Hochberg)、"BY"(Benjamini-Yekutieli)、"bonferroni"等。
pvalueCutoff = 0.05, # 用于筛选显著富集项的p值阈值。
qvalueCutoff = 0.2) # 筛选显著富集项的校正后p值(q值)阈值。
# order:指定结果排序的依据,如"p.adjust"(校正后的p值)、"p.value"(原始p值)等。
# 查看富集分析结果
head(enrich_result)
# 查看富集分析结果的摘要
summary(enrich_result)
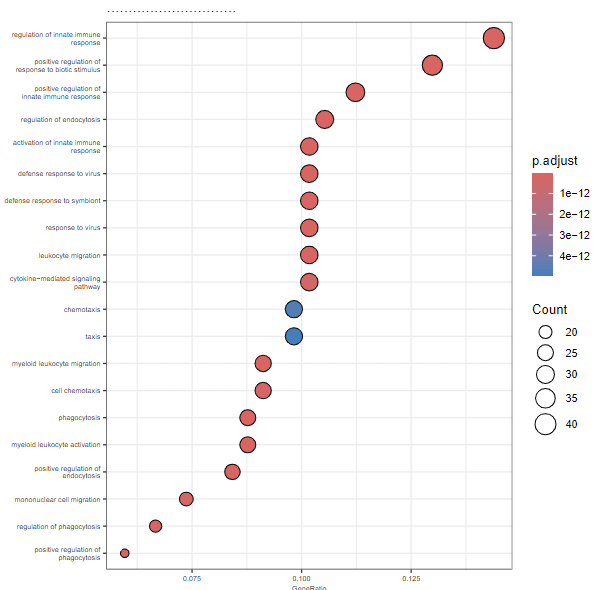
dotplot(enrich_result, showCategory = 20,font.size = 6)
}
出图