官网API: https://scirpy.scverse.org/en/latest/index.html
加载数据
# 定义类型Type
cluster_to_celltype = {
0: 'CD8_Teff-GZMK',
1: 'CD8_Teff-GZMK', # 和0一样
2: 'CD4_Treg',
4: 'CD4_TN',
5: 'NKT',
6: 'CD8_Tcm',
7: 'CD8_Teff-CD160',
8: 'CD8_Tex-LAYN',
9: 'CD8_Tex-LAYN', # 8和9一样
10: 'CD4_Tcm',
11: 'CD8_Tpro-MCM7',
12: 'yðT',
13: 'CD8_Tex-XCL2',
14: 'CD8_Tpro-MKI67',
15: 'CD8_Tex-MX1',
16: 'CD8_Teff-CD160', # 16和7一样
17: 'CD8_Tex-HSP'
}
h5file = "/home/data/TCR_BCR_Data/Anno.h5ad" # 设置Seurat对象位置
adata_gex = sc.read_h5ad(h5file)
# 将数据洗成需要的格式
if 'seurat_clusters' in adata_gex.obs:
# 使用map函数映射新的celltype列
adata_gex.obs['celltype'] = adata_gex.obs['seurat_clusters'].map(cluster_to_celltype)
# 读取免疫组库数据
adata = ir.io.read_10x_vdj("vdj_v1_hs_pbmc2_t_filtered_contig_annotations.csv")
# 数据预处理
# 我们用muon来来整理数据格式
mdata = mu.MuData({"gex": adata_gex, "airr": adata})
ir.pp.index_chains(mdata)
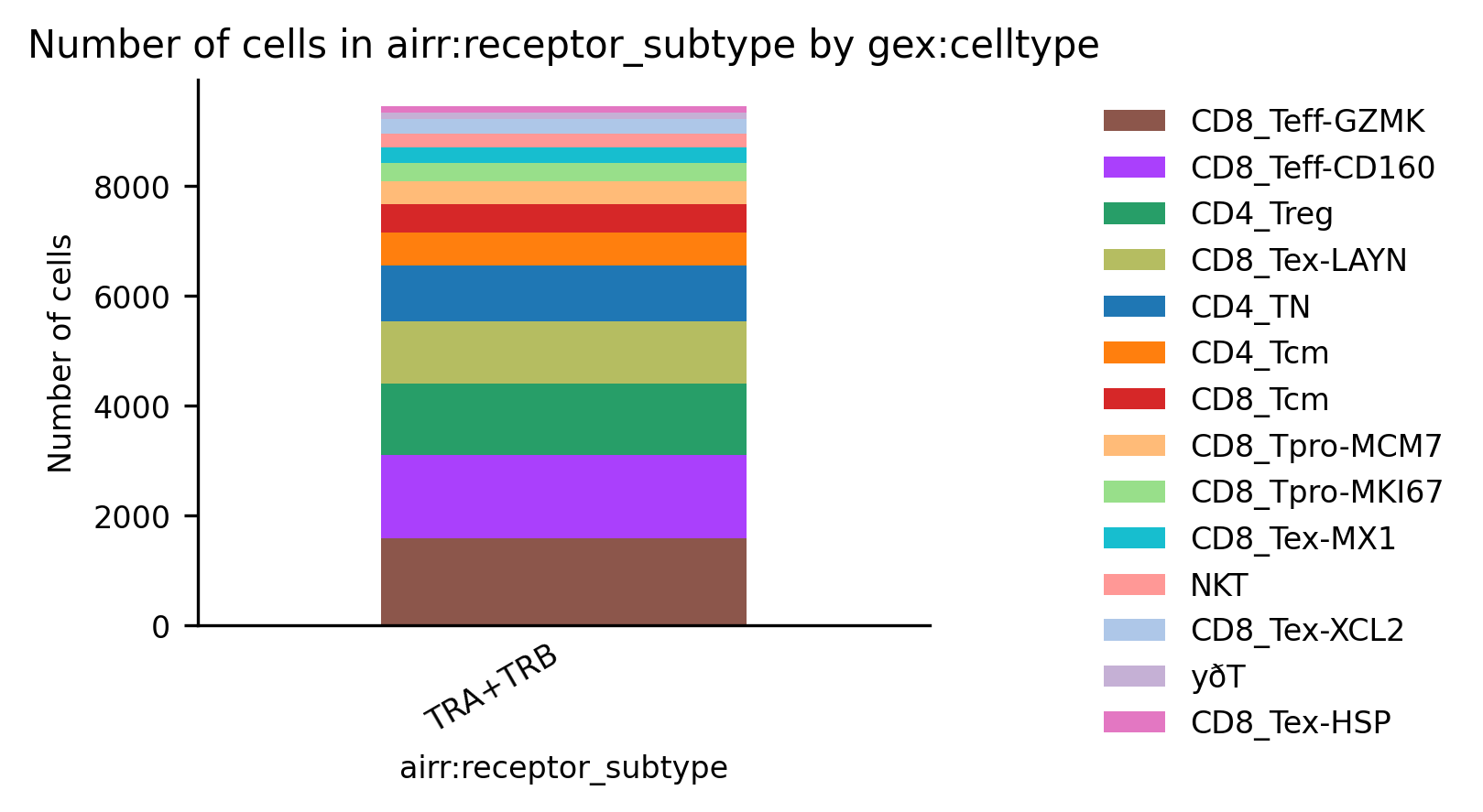
# 标记免疫细胞受体组成,可以使用 scirpy.pl.group_abundance 实现可视化。
ir.tl.chain_qc(mdata)
ir.pl.group_abundance(mdata, groupby="airr:receptor_subtype", target_col=celltypecol)
ir.pl.group_abundance(mdata, groupby="airr:chain_pairing", target_col=celltypecol)
receptor_type是指粗略分类为BCR和TCR。 同时具有“BCR”和“TCR”链的细胞被标记为“ambiguous”。receptor_subtype是指更具体的分类,分为 α/β、ɣ/δ、IG-λ 和 IG-κ 链配置。
Stored result in `mdata.obs["airr:receptor_type"]`.
Stored result in `mdata.obs["airr:receptor_subtype"]`.
Stored result in `mdata.obs["airr:chain_pairing"]`.
# multichain细胞和不具有至少一对完整受体序列的细胞都应从下游分析中排除
mu.pp.filter_obs(mdata, "airr:chain_pairing", lambda x: x != "multichain")
mu.pp.filter_obs(mdata, "airr:chain_pairing", lambda x: ~np.isin(x, ["orphan VDJ", "orphan VJ"]))
# 在经过质控后再次进行分布可视化
ir.pl.group_abundance(mdata, groupby="airr:chain_pairing", target_col=celltypecol)
克隆型分析
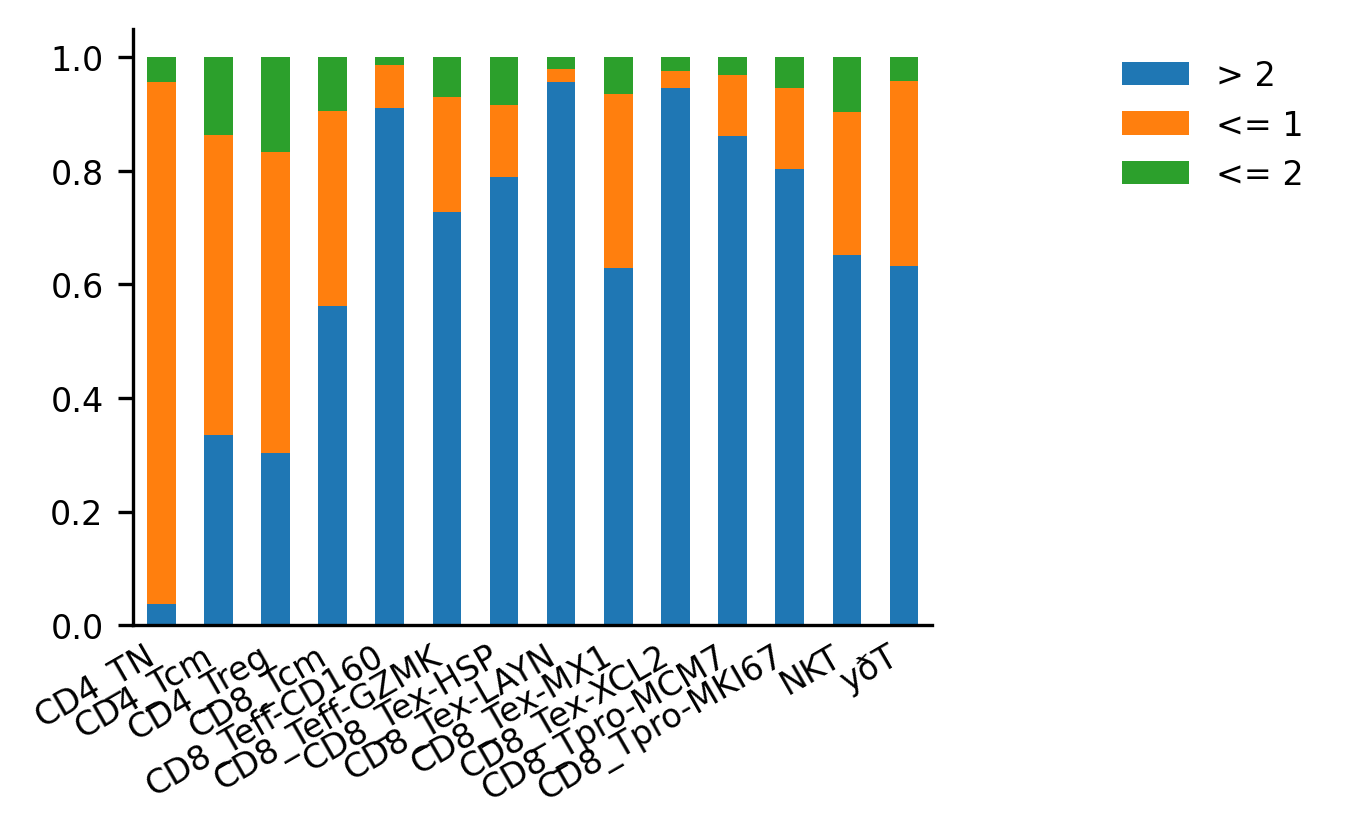
- 克隆扩增
在特定生物学条件或治疗反应中,某些克隆可能会发生扩展。通过追踪这些克隆的变化,可以更好地理解免疫应答的动态变化。
# 计算克隆扩展
ir.tl.clonal_expansion(mdata, groupby='condition')
# 可视化克隆扩展状态
ir.pl.clonal_expansion(mdata, target_col="clone_id", groupby=celltypecol, clip_at=4, normalize=False)
克隆型多样性可以通过计算多样性指数来评估,如 Shannon 多样性指数或 Simpson 指数。这些指数能帮助我们了解样本中克隆的丰富度和均匀度。
# 计算 Shannon 多样性指数
ir.tl.alpha_diversity(mdata, metric="shannon", groupby='sample', target_col='alpha_diversity_shannon')
# 计算 Simpson 多样性指数
ir.tl.alpha_diversity(mdata, metric="simpson", groupby='sample', target_col='alpha_diversity_simpson')
# 可视化结果
sc.pl.violin(mdata, ['alpha_diversity_shannon', 'alpha_diversity_simpson'], groupby='sample')
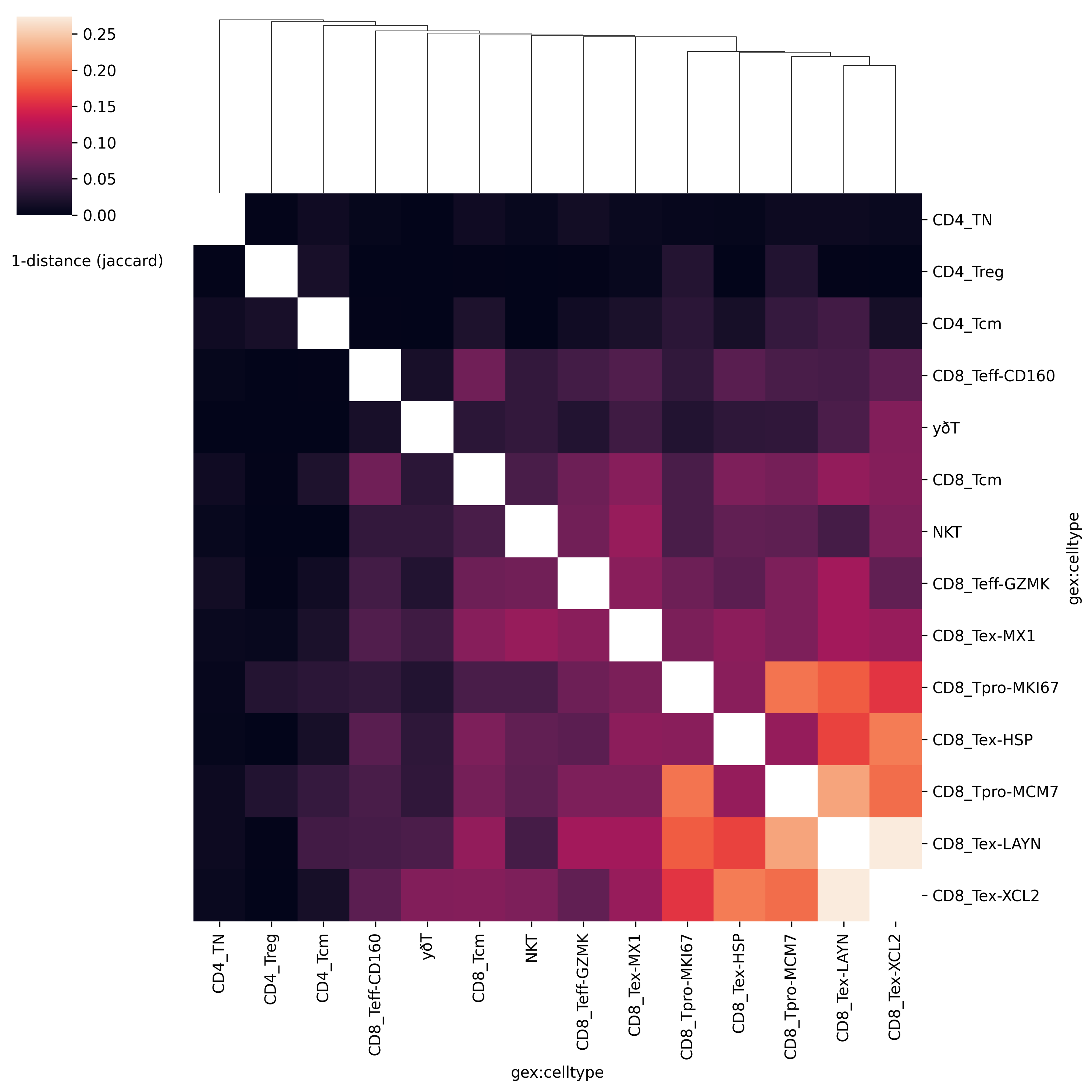
Repertoire Overlap 分析
Repertoire overlap 分析用于计算样本之间的克隆重叠度,这有助于了解不同样本(例如来自不同时间点或不同治疗组的样本)之间的相似性。
在Python中,可以使用scirpy库来模拟R语言的immunarch包中的repOverlap功能。immunarch的repOverlap方法用于计算不同样本之间的受体重叠度,这在研究免疫应答中的相似性和差异性时非常有用。在scirpy中,可以使用ir.tl.repertoire_overlap来实现类似的功能。这个方法计算了样本之间共有的克隆数量,并可以用于比较免疫库的相似性。
# 计算重叠度
ir.tl.repertoire_overlap(mdata, groupby=celltypecol)
print(mdata.obs.columns)
# 绘制热力图
ir.pl.repertoire_overlap(mdata, groupby=celltypecol, cmap="viridis")
plt.savefig("repertoire_overlap_E.png", dpi=300, bbox_inches='tight')
这会生成一个热图,显示不同样本间的重叠度。重叠度较高可能表明相似的免疫反应或共享的免疫记忆。
这两种分析方法提供了对 TCR/BCR 克隆多样性和相似性的量化和可视化,有助于理解免疫系统的动态变化和功能。